 Frequently Asked Questions
Frequently Asked Questions
Is there a FROGS Guidelines or standard procedure?
FROGS' design is highly modular and so allows users to choose their tools and processing order. However, default values are advised when possible, and a standard procedure for amplicon analysis should follow these steps:- Pre-processing: depending on your data, assemble or not your paired-reads (2 reads will be merged if they overlap at least on 10 bases). Depending on your studied amplicon, fill the size parameters and primers fields.
- Clustering: thanks to Swarm capacities, clustering can be performed early in the process. It should be performed with an aggregation distance of 3 and with a denoising step.
- Removing chimeras: chimeras are PCR artifacts and should be removed at this step, using the clusters produced by Swarm.
- Filtering OTUs: a 0.005% abundance threshold should be apply to remove the remaining noisy clusters and obtain your OTUs. If your experimental design contain replicates, you should also filter clusters which are not present in at least two/three/more samples (depending on your design).
- ITSx (optional): If you analysed fungi ITS you can choose to keep only sequence with ITS signature, in this case, you have to use ITSx (see belon How to use FROGS to analyse ITS reads ? section)
- Taxonomic affiliation: this step should be executed at the end of the process because it is the most time consuming one. Default is to produce blast affiliation and multi-affiliations, but RDP affiliation can be added.
- Visualization (optional): use “Cluster stat” (after steps 2, 3 and 4) for some supplementary figures and stats about your clusters (numbers, distributions etc.). Use “Affiliations stat” for some supplementary figures and stats (after step 5)
- Tree construction (optional): use it after step 4) if you want a phylogenetic tree of your OTUs
- Export functions (optional): use “BIOM to TSV” if you want an abundance table in tabular format. Use “BIOM to standard BIOM” if you need a BIOM file for your statistical analyses.
What data are processed by FROGS?
FROGS is designed for the study of microbial communities from amplicon sequencing. The amplified area is chosen to be as distinctive as possible in the community you are interested in. For example, researchers favour 16S ribosomal RNA part when studying the bacterial composition of an environment.However, FROGS can be used on any amplicon as long as the area of interest respects the constraints mentioned below:
- FROGS works on ribosomic RNA 16S, 18S, 23S, but also amplicons belonging from functionnal genes such as dsrB. FROGS can also analysed ITS reads since FROGS v3.0 and more broadly, reads with hypervariable amplicon lengths that may not have overlapping read pairs. The only limit is the reference database provided to the software.
- FROGS has been designed to manage data from Illumina sequencers (MiniSeq, MiSeq, NextSeq, HiSeq,...). It can, however, accept data from 454 if it is in fastq format. If your data is in SFF format you can use the SFF converter software to make fastq data (don't forget to use the option to remove adapter 454 and low quality sequences).
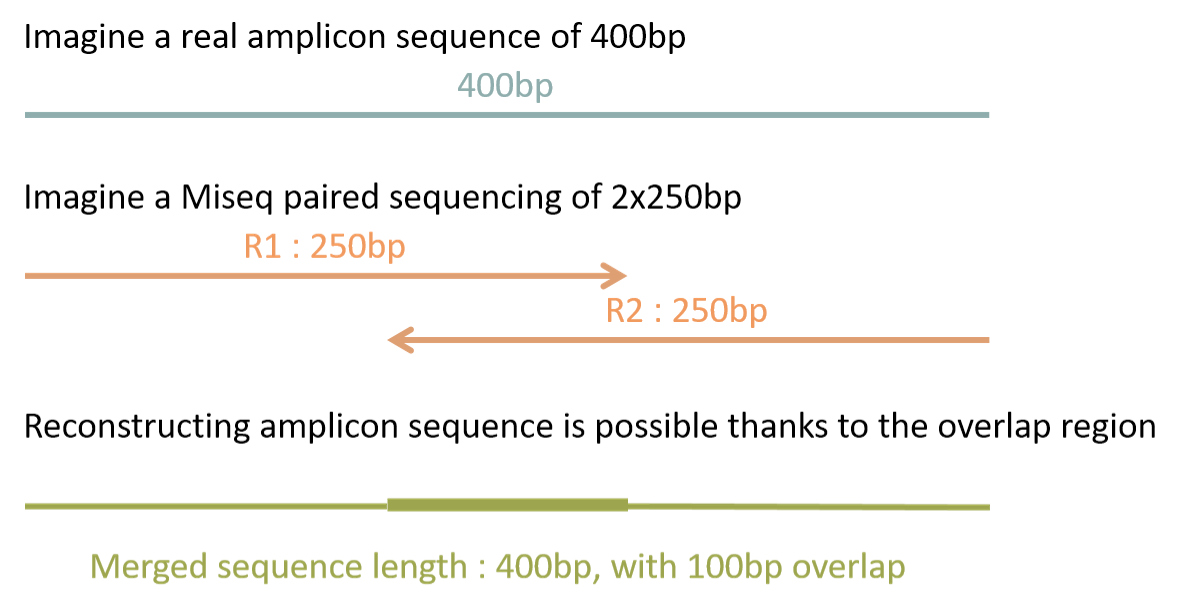
- The diagram below shows the main sequencing modes supported or not supported by FROGS:

In standard protocol, target DNA must be completely sequenced in the reads i.e. either a single-end reads starting from 5’ primer and finishing to the end of the 3’ primer or in paired-end case the forward and reverse reads must be overlapped.
What primers can I used for my amplicons ?
Exemples for 16S Bacteria:
http://help.ezbiocloud.net/16s-rrna-and-16s-rrna-geneExemples for Fungi and other amplicons:
http://www.fungalbarcoding.org/DefaultInfo.aspx?Page=PrimersHow to use FROGS to analyse ITS reads ?
Preprocess Tool
ITS reads can have non-overlapping reads. Merging tool as VSEARCH cannot merge these reads and they are lost by the system (two reads will be merged if they overlap at least on 10 bases). So, to keep these reads we created an option on Preprocess Tool, activated in replying "Yes" to "Would you like to keep unmerged reads?"These sequenced are tagged combined sequences.

Carreful, this option have to be activated only if reads are longer than target sequence. Otherwise, you will keep noise in your analyses. the "un-merged" will be "combined" sequences.
Difference between "overlapped" and "combined" sequences:

FROGS "combined" sequences are artificial and present particular features especially on size.
Imagine a MiSeq sequencing of 2x250pb with reads impossible to overlap. So FROGS "combined" length = 600 bp.
What is the purpose of the ITSx tool?
ITSx is a tool to filter sequences. ITSx identifies and trimms ITS regions in our sequences. It excludes the highly conserved neighbouring sequences SSU, 5S and ARNr LSU (see figure below). If the ITS1 or ITS2 region is not detected, the sequence is discarded. You can choose to check only if the sequence is detected as an ITS. In this case, the sequence is not trimmed, only sequences not detected as ITS are rejected (e.g. contaminants).
It is interesting to keep only the ITS parts without the flanking sequences in case one would like to compare sequenced amplicons with different primers targeting the same region to be amplified. You can choose this option on configuration panel of ITSx Tool. Reply "No" to question "Check only if sequence is detected as ITS?". In opposite, if "Yes" is chosen, sequences with ITS signature will be kept without trimming SSU, LSU or 5.8S regions.

Carreful, The ITSx step is time consumming and has to be done on clusters. We advise our users to apply ITSx in 5th step:
- Preprocess step,
- Clustering step,
- Chimera removing step,
- Filter on OTUs abundances and replicats step,
- ITSx if Fungi ITS amplicons.
Carreful, ITSx is currently usable for the detection of fungi ITS (neither plants nor other eukaryotes).
What is special about the affiliation of ITS (with combined sequences more broadly)?
blastn+ or needleall is used to find alignment between each OTU and the database. Only the bests hits with the same score are reported. blastn+ is used for merged read pair, and needall is used for artificially combined sequence. For each alignment returned, several metrics are computed: identity percentage, coverage percentage, and alignment length. If "combined" sequences are stayed presents in OTUs, blastn+ is not usable as for classical merged sequences. So, sequences are affiliated in 3 steps:- Alignment of classical "merged" sequences with blastn+ versus chosen database (e.g. UNITE),
- Alignment of "combined" sequences with blastn+ versus chosen database, best hits are collected and a very small new databank (at most 200 references per blast hit) is created composed exclusively of "subject" sequences from these best hits,
- Alignment of "combined" sequences with needleall (global alignment: very time consumming) versus these small new databank.
Carreful, with "combined" sequences, we introduced some modification on identity percentage



This calculation allows the 100% identity score to be returned on FROGS "combined" shorter or longer than reality in case of perfect sequencing. And returns a lower percentage of identity in the case of repeated small overlaps kept in the FROGS "combined".
What is the purpose of the Affiliation post-process tool (since FROGS v3.0)?
This tool allows grouping OTUs together in accordance with the %id and %cov chosen by the user and according to the following criteria:- They must have the same affiliation Or
- If they have "multi-affiliation" tag in FROGS taxonomy, they must have in common in their list of possible affiliations at least one identical affiliation.
- The different affiliations involved in multi-affiliation are merged.
- The abundances are added together.
- It is the most abundant OTU seed that is preserved.

What filters should I use in FROGS-Filters?
FROGS proposes various filters to meet users'needs, but depending on your data and scientific question, only a part of them should be used. The most used filters are the OTU filters based on samples and abundances.- The “minimum number of samples” parameter should be used when processing datasets with replicated samples, or repeated samples. Except if you are particularly interested in OTUs that are not shared between your replicates or repetitions, this parameter allows you to remove all OTUs that are not detected in at least X samples (X corresponding to your level of replication/repetition).
- The “minimum proportion/number” parameter should always be used unless you are focusing on extremely rare OTUs and don’t care about false positive OTUs. Indeed, to ensure a good community description, a 0.005% abundance filter should be apply to your data.
- The “N biggest OTU” parameter is available if you have any reason to focus only on the biggest OTUs and want to remove the smallest ones. It should be use only with well-known samples or when interested in specific major OTUs.
- The taxonomic filters should be used only in specific situations, with well-known communities when not interested in poorly characterized taxa, or if you are interested only in some chosen and known taxa. They allow you to filter OTUs with bad taxonomic affiliations, based on thresholds applied to RDP bootstraps, blast e-value, identity or coverage.
- Finally, the contamination filter allow you to remove known contaminants, using a user-provided list. Remaining phiX sequences, technical artifacts introduced during Illumina could also be filtered with this filter.
How to fill the 5’ and 3’ primers fields in FROGS-Preprocess tool?
The most frequent error encountered by FROGS new users is a wrong completion of their 5’ and 3’ primers. Make sure that you followed the instructions available at the bottom of the tool: primers should be provided as they are read on a 5’->3’ sequence. Generally, that means that your 3’ primers should be reverse-transcripted.Example:
5' ATGCCC GTCGTCGTAAAATGC ATTTCAG 3'Value for parameter 5' primer: ATGCC
Value for parameter 3' primer: ATTTCAG
Degenerated nucleotides are accepted.
What is the “custom protocol” parameter in FROGS-Preprocess tool?
(Illumina data only) This custom protocol corresponds to Kozich et al. (2013) protocol, where PCR primers are also used as sequencing adaptors, and so PCR primers are not included in the obtained sequences. Thus, choose this parameter only if you used such a sequencing protocol, otherwise, select the standard protocol.What is the “mismatch rate” parameter in FROGS-Preprocess tool?
(paired reads Illumina only) The mismatch rate corresponds to the mismatch rate allowed when pairing paired-end reads using Flash in the FROGS-Preprocess tool. A 0.1 rate means that 10% mismatches are allowed along the overlapping region. Sequences presenting more mismatches will be discarded.What are the “minimum/maximum amplicon size” parameters in FROGS-Preprocess tool?
(paired reads Illumina only) These parameters correspond to the sizes below/above which assembled paired reads will be discarded. They allow to filter badly assembled paired reads.What is Swarm and how does it work?
Swarm is a novel clustering algorithm which does not rely on a fixed global clustering threshold. For more information about Swarm, consult https://github.com/torognes/swarm or Mahé F, et al. (2014) and Mahé F, et al. (2015)Who uses FROGS?
Dimensions statisticsGoogle Scholar statistics